-

© iStock.com/carloscastilla

Research Projects
Comparitive analysis of small RNA regulatory networks in Gammaproteobacteria
Muhammad Elhossary (Supervisors: Förstner, Stoye)

In this project, we aim to identify, annotate and characterize novel sRNAs from a diverse set of microbes from the class of Gammaproteobacteria (a total of 20 species). Samples will be collected from four different growth conditions including iron-limitation and cell membrane stress to ensure that sRNAs expressed under a broad range of environmental settings will be detected. Focusing on sRNA regulators that depend on the RNA chaperon Hfq for their function and act through base-pairing with target mRNAs, we will study their distribution as well as their evolution.
The figure shows an example of small RNA regulatory network,
source: Wagner and Romby 2015
Characterizing biogas microbioms by meta analysis of metagenomes
Benedikt Osterholz (Supervisors: Sczyrba, Schlüter)
In anaerobic digestion of biomass, a huge number of microbial species is involved possessing a wide variety of metabolic properties. However, a major part of the species that can be detected in biogas reactors has not been adequately characterized either in terms of its specific substance conversion properties or in terms of its respective ecological role in the microbiological system. Accordingly, the trophic network responsible for the degradation of crop biomass in biogas reactors is understood to be only piecemeal and only in terms of basic microbial processes.
The aim of this project is the use of high-throughput molecular data for a detailed representation of microbial networks by means of a comprehensive bioinformatic evaluation (meta-analysis) including abiotic process factors. Established bioinformatics solutions and concepts will be reimplemented to make optimal use of available de.NBI cloud resources to identify the core-microbiome of biogas communities, determine unique taxa for specific communities and elucidate relationships between taxonomic units. It is expected that obtained results will contribute to the identification and characterization of key organisms to better understand and improve the biogas process at a whole.
Software for computational pangenomics
Andreas Rempel (Supervisors: Stoye, Förstner)

A pangenome is a collection of genomic sequences from different individuals. It holds information on conserved regions, local polymorphisms, and structural variations and can provide insights into genomic differences and evolutionary relationships. There are different data structures used for the storage and comparison of the sequences, such as colored De Bruijn Graphs, Variation Graphs, or Sequence Bloom Trees. The aim of this project is to compare existing software tools for computational pangenomics, to define a common standard interface for the data structures, and to set up an automated (cloud-based) test environment to evaluate their performance and to support users in finding the tool that suits their demands best.
Bioinformatics solutions for microbiome meta-transcriptome analyses
Tom Tubbesing (Supervisors: Sczyrba, Schlüter)
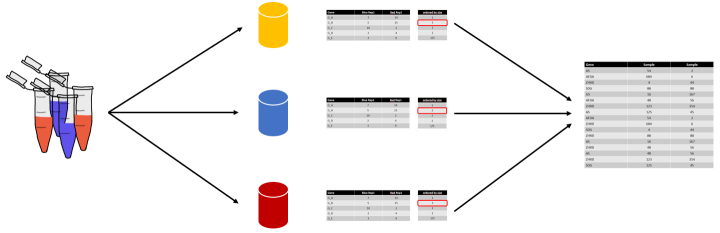
Analysing the transcriptome of microbial strains to identify Differentially Expressed Genes (DEGs) is a common approach. The DESeq2-package (Love et al., 2014) is well established for carrying out this kind of analyses based on count data from RNA sequencing experiments. However, when studying microbial communities, reliably identifying DEGs based on a metatranscriptome sequencing datasets is compounded by the fact that the abundances of microbial taxa vary between sampling conditions. This project is aimed at implementing a comprehensive software workflow for the analysis of such datasets and use it to expand the Elastic MetaGenome Browser (EMGB) platform.
Modeling of biological networks and development of user-friendly software for modeling applications
Emanuel Lange (Supervisors: Heyer, Nattkemper)
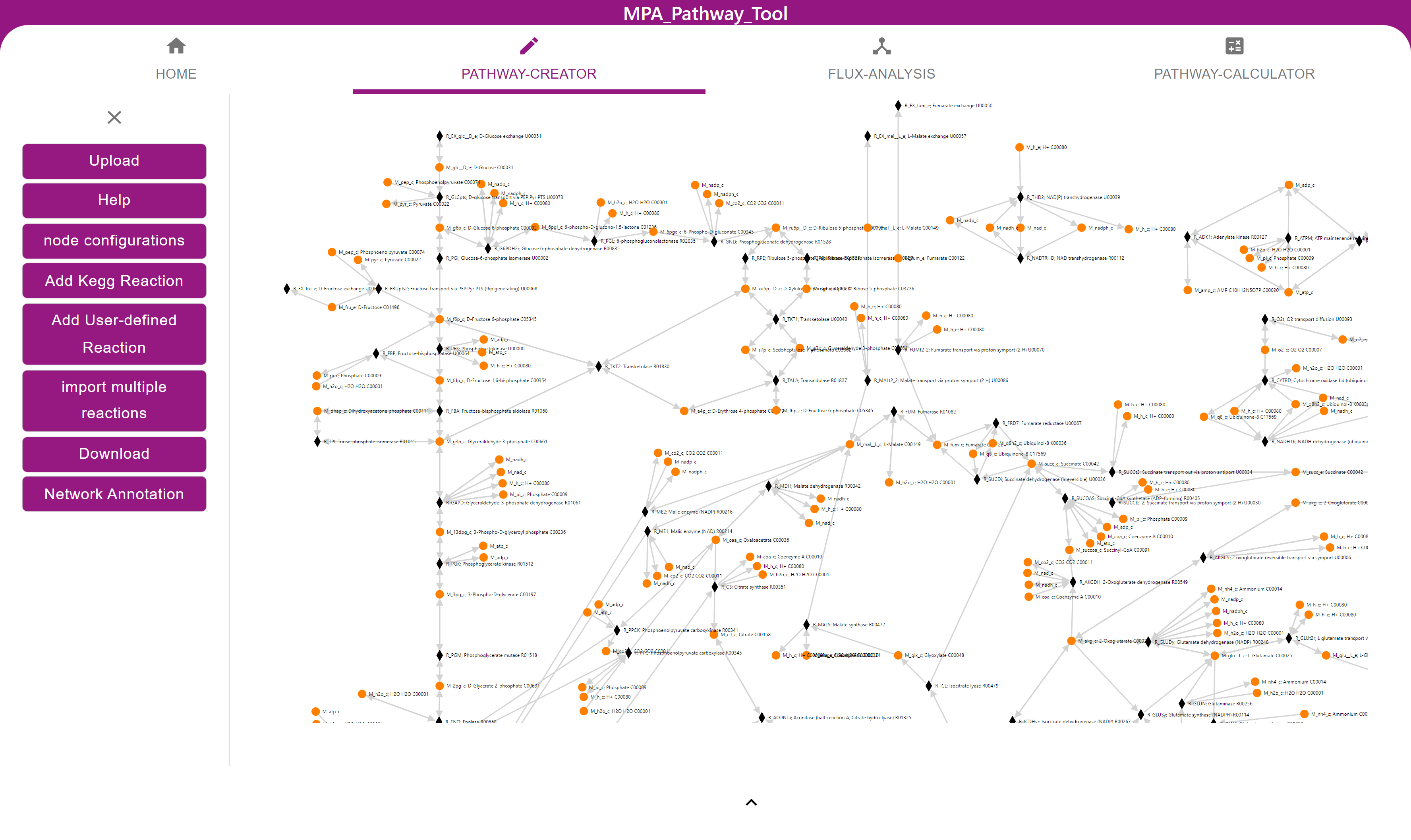
Physiological processes in living cells are controlled by metabolism, signaling, and regulatory networks. Existing knowledge of these biological networks can be compiled into mechanistic models, which facilitate comprehension, prediction, and optimization of cellular processes. Model predictions can be improved, by incorporating omics data into them. At the same time, analysis and interpretation of omics data can benefit from model predictions. However, models are rarely used in experimental studies generating omics data.
The first objective of my project is to develop strategies to integrate models and omics data with two potential applications: The investigation of cancer metabolism, and the investigation of signaling in neutrophil granulocyte migration. My second objective is to make modeling more accessible for experimentalists to establish models for experimental studies. To achieve this goal, I plan to implement user-friendly capabilities for modeling, data analysis, and algorithms for network visualization into our “MPA-Pathway-Tool” (Walke et al., 2021). The “MPA-Pathway-Tool” is a web application already supporting pathway mapping and metabolic modeling.
Conversion of molecular passport data for phylogenetic analysis and accession selection
Manuel Feser (Supervisors: Scholz, Sczyrba)
The main objective of this project is to convert the molecular passport data (diversity matrix) of selected genebank accessions (Plant Genetic Resources) into a data structure that can be stored and used for analyses. For example, a user may have a diversity vector of a genotype of interest and wishes to find the phylogenetically closest genebank material from a particular geographical region or with particular traits. To increase the power of the analysis, an imputation service called DivImpute is developed. This will increase the marker density and enrich the input for the subsequent phylogenetic similarity search. DivImpute is designed as a cloud-enabled pipeline, minimizing the cost of imputation by distributing the computational load, with the input split into overlapping genome windows.
Comparative Pangenomics
Leonard Bohnenkämper (Supervisors: Stoye, Bräutigam)
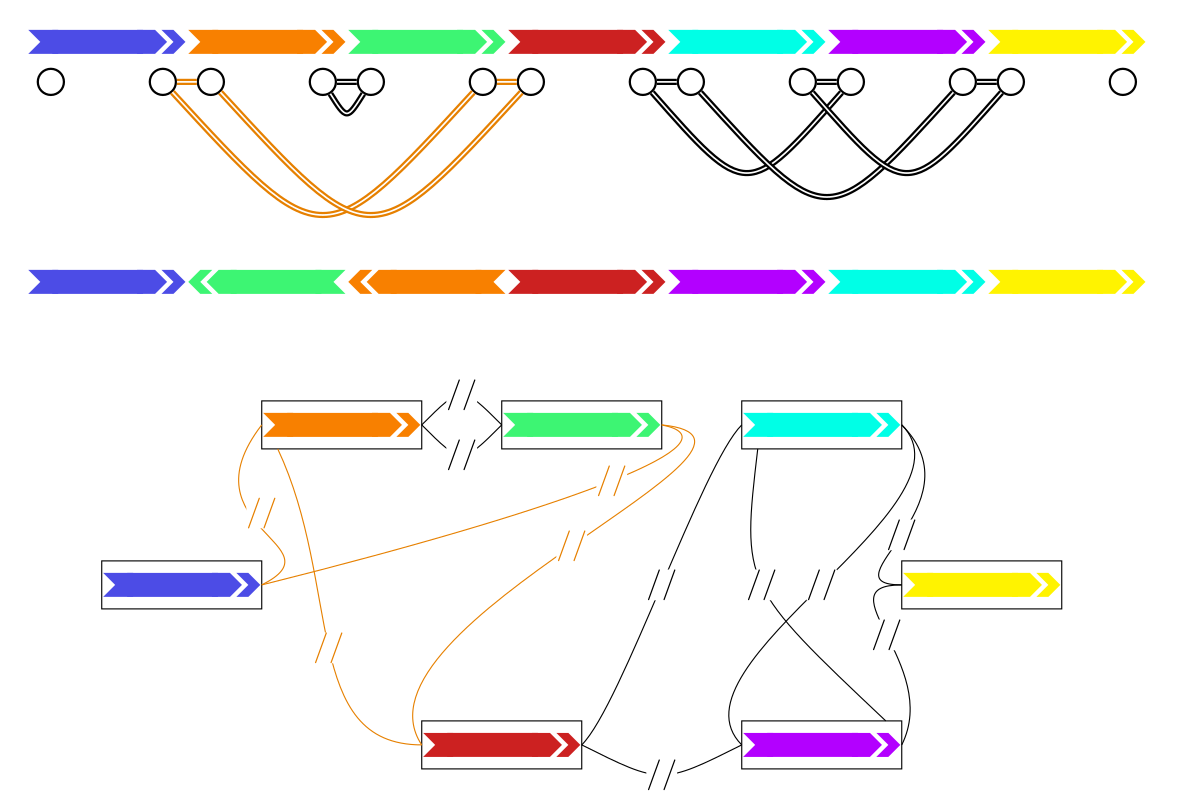
Genome rearrangements have been studied extensively in theoretical works of Comparative Genomics. These results however, have only been applied on a limited scale to real genomes. The continuing progress of sequencing projects and technology made more and more high quality genomes available and enabled even Pangenomic analyses, that is, analyses that include all availailable genomes of a species. Pangenomics and theoretical Rearrangement Studies utilize remarkably similar graph data structures. Given the abundance of theoretical results in Comparative Genomics, it is likely that many of these results can be applied in Pangenomics. Conversely, the abundance of practical results in the construction of Pangenome graphs can likely contribute to these theoretical results seeing more real world applications.
Graph Neural Networks and Explainable AI for a new Aurora kinase inhibitor
Luna Pianesi (Supervisor: Schönhuth)
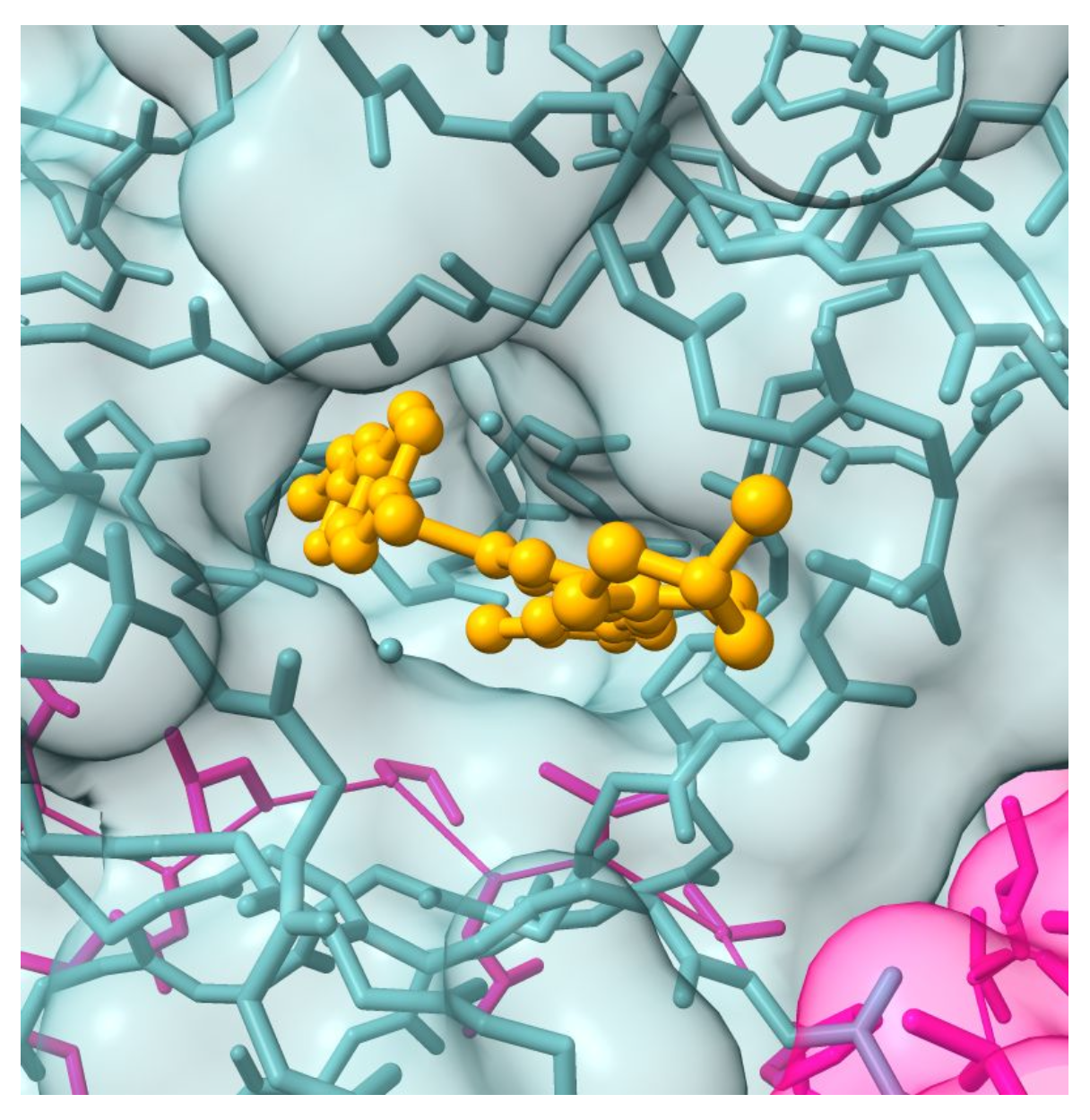
Drug Discovery has long needed a speed-up of some sort in its process, and computational drug design might come to the rescue. By exploiting the vast potential of artificial neural networks - and particularly Graph Neural Networks - one could devise a fully computational drug design pipeline to address the huge work that goes into pre-clinical studies. Developing fruitful partnerships between artificial intelligence and biology can lead to a flexible method that is able to produce hundreds of novel drug candidates targeting a large variety of biological targets. This study currently focuses on de novo design of ATP-competitive small-molecule inhibitors for the cancer-inducing dysregulation of the Aurora protein kinase.
Construction and Analysis of Knowledge Graph Representation of Bacterial small RNAs and their Interactions
Vanessa Scharf (Supervisors: Förstner, Cimiano)
Bacterial small non-coding RNAs are important molecular regulators, and help bacteria as well as archaea to adapt efficiently to environmental conditions. Besides other regulators, they are part of complex molecular networks in those species but are often neglected in the modelling of those. Their detection and characterization as well as the description of interaction partners (other RNAs or proteins) is very costly and the results of such studies are often only deposited in small data bases or are only found individually in scholarly articles. This project aims to model sRNAs and their features as well at the interaction topology in an open knowledge graph. This will make the knowledge about these regulators sustainably accessible, easily query-able and connected to further databases. Having the knowledge of these regulators of several species in such a machine readable form and linked to other databases will enable to apply graph machine learning approaches and, by that, to perform link prediction and further analysis methods. With this approach, the project aims to conduct network inference and to reveal so far unknown regulatory interactions while also making information about sRNAs efficiently searchable and accessible.
Potential of Knowledge Graphs for Proteomics Research
Daniel Kautzner (Supervisors: Heyer, Schönhuth)
Knowledge graphs are emerging as a powerful tool in biomedical and clinical research, offering a structured framework to integrate and analyze complex biological datasets. By connecting diverse data sources, they facilitate a deeper and more comprehensive understanding of biomedical information.
The primary objective of this project is to evaluate the suitability of existing knowledge graphs for proteomics research, emphasizing testing their capabilities using real-world patient datasets. Key areas of exploration include their potential for applications such as disease prediction and drug recommendation, both of which are critical for advancing precision medicine. The project also aims to address challenges related to accessibility and usability by developing automated processes for personalized applications and creating intuitive tools to support lab researchers. While some knowledge graphs are available for human proteomics, they are limited, and there is a notable lack of freely accessible options for metaproteomics or other specialized proteomics fields. This gap will be addressed as part of this work, which focuses on adapting and optimizing knowledge graphs for metaproteomics, facilitating more in-depth analyses of protein communities within complex biological systems.
MetaProtPanel: Development of a fecal metaproteomics panel by crossvalidation of protein biomarkers
Maximilian Wolf (Supervisors: Heyer, Sczyrba)
The human gastrointestinal tract hosts a vast and diverse community of microbial species, which play essential roles in health and disease by, for example, contributing to food digestion and modulating immune responses. Metaproteomics offers a means to characterize interactions between the human host and microbiome by identifying and quantifying microbial and host proteins present in complex biological samples. This approach enables the detection of key organisms or functional pathways that may be disrupted in diseases such as inflammatory bowel disease (IBD) or colorectal cancer. However, metaproteomic studies face several challenges, including variability in wet-lab procedures and bioinformatics workflows, which can lead to discrepancies and contrasting findings across studies. The goal of this project is to develop and implement a comprehensive workflow to facilitate the consistent processing and analysis of metaproteomic data from repositories, utilizing state-of-the-art metaproteomic and proteogenomic methodologies as well as advanced data processing and analysis strategies. This integrated approach will support a meta-analysis of metaproteomic datasets and aid in identifying universal protein biomarkers of human and microbial origin that are associated with diseases like IBD.
Boolean modelling of signal transduction: integrating knowledge graphs and phosphoproteomics, and the development of a user-friendly modelling tool.
Benjamin Saalfeld (Supervisors: Heyer, Lutter)
Biomolecules form complex interacting systems at various biological levels, where the dynamics of their interactions drive cellular phenotypes and responses. Signal transduction and gene regulatory networks serve as typical examples of these biological systems. Boolean models are employed to characterize the entities within such systems, using binary state variables and rules that define their influence on one another, notably without requiring detailed kinetic or quantitative information. The goal of this project is to advance the Boolean modeling of signal transduction pathways, with a particular focus on leveraging phosphoproteomics data. A central component of this research is the development of a dedicated modeling tool. The core methodology will involve the integration of existing literature-derived biological knowledge into knowledge graphs, utilizing the Neo4j graph database. This approach is designed to facilitate the direct generation of Boolean models from these structured knowledge graphs, with the flexibility to either incorporate or exclude phosphoproteomics data during model construction. The practical utility and effectiveness of the developed methodology and tool will be demonstrated through its application to two specific biological use cases: the activation of platelets and the activation of neutrophils.
Completed PhD projects
Measures and algorithms for alignment-free comparative pan-genomics
Luca Parmigiani (Supervisor: Stoye)

Pangenomics aims to capture the collective genomic diversity of taxonomically related genomes, typically from the same species, offering potential insights for medicine and biology. Initially defined as collections of genes, pangenomes are now more accurately represented through variation graphs. These graphs illustrate genomes by using nodes to depict shared sequences, edges to denote sequential connections, and paths to reconstruct the original genomes. Despite their benefits, there is a lack of scalable software for essential analyses, such as estimating the core genome size and evaluating the extent of genomic variability (pangenome growth). To address this need, we developed Panacus (pangenome-abacus), a tool designed for efficient data extraction from GFA files, the standard for pangenome graphs. Panacus facilitates quick generation of pangenome growth and core curves, handling millions of nodes in variation graphs within minutes.
Functional genomics of and bioinformatic analyses tools for seed quality parameters in rapeseed
Hanna Marie Schilbert (Supervisors: Weisshaar, Holtgräwe, Stoye)
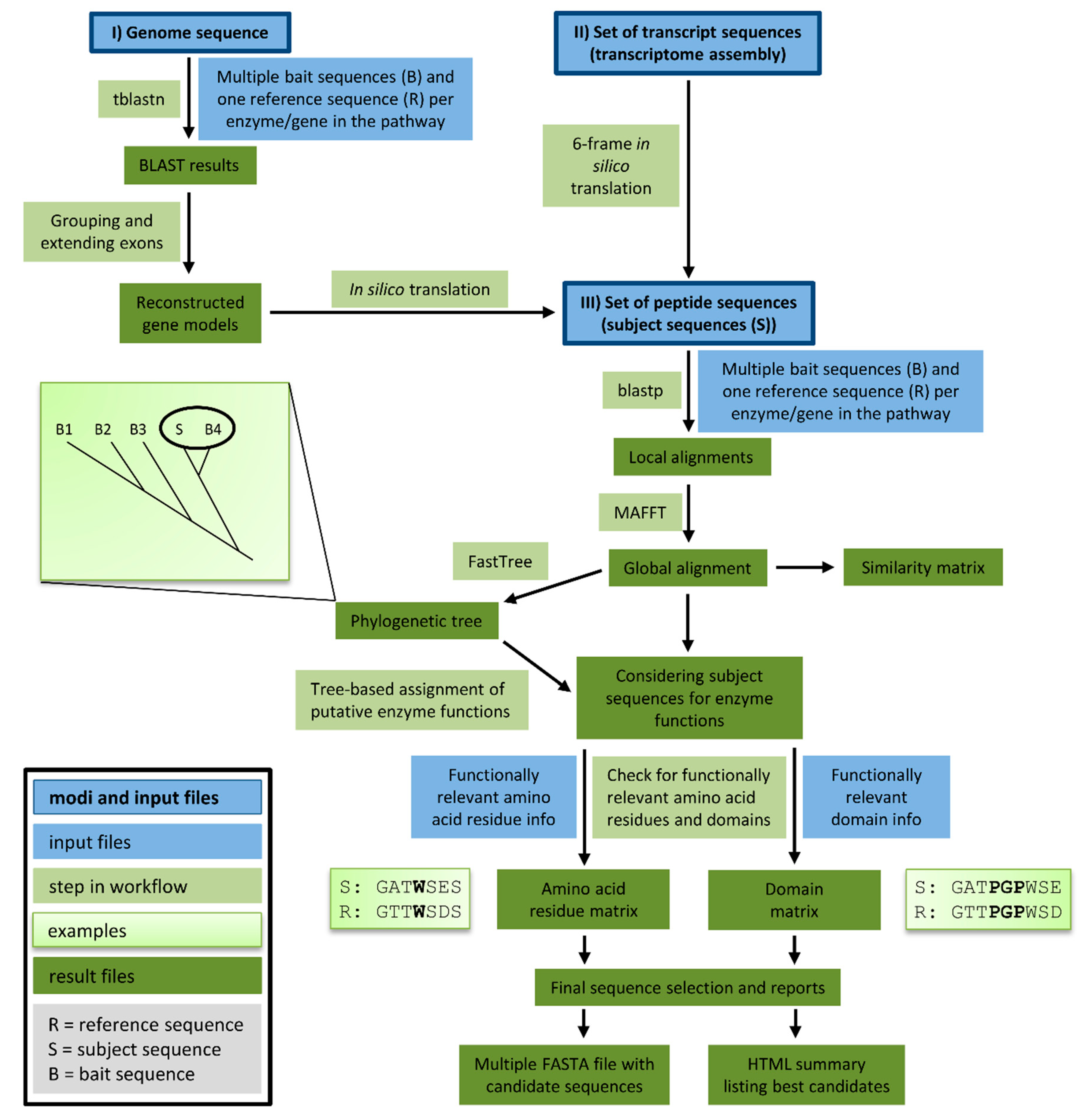
The increasing demand in high quality plant based food products requires the generation of improved crops. To achieve this aim, the molecular basis of relevant seed quality parameters needs to be analysed in relevant crop species. By harnessing large genomic and transcriptomic data sets, loci and genes associated with seed oil-, seed protein-, and antinutrients content will be identified in rapeseed (Brassica napus). The development of dedicated tools will help to facilitate the automatic analyses of involved genes and encoded enzymes and provide predictions for their functionalities. Tools will be made freely available on github, e.g. KIPEs (Knowledge-Based Identification of Pathway Enzymes).
Figure source: Pucker, Reiher, Schilbert 2020

© shutterstock.com/pingebat
From hidden data and information towards data-driven research
Lisa Kühnel (Supervisors: Fluck, Cimiano)
The enormous growth in electronic research data requires semantic interoperability and computational methods to generate information and knowledge. However, heterogeneity, restricted access, non-standardized and low-quality data hamper data-driven research in the (bio)medical area. This thesis investigates the application of computational methods to convert (bio)medical data and information into accessible, machine-readable formats with the aim to support researchers. Thereby, the focus lies on two data types: 1) For biomedical literature, this work investigates the robustness of state-of-the-art NLP methods to allow the transfer from science to services. 2) For clinical data, the reliability of synthetic data generation algorithms based on a defined use case is examined.
Machine readability and access to data, information and knowledge are core requirements for data-driven research. Furthermore, the enormous growth in freely available, electronic research data increases the need for semantic interoperability as well as computational methods to generate new information and knowledge from the data.
Applications of colored de Bruijn graphs
Tizian Schulz (Supervisors: Stoye, Hach)
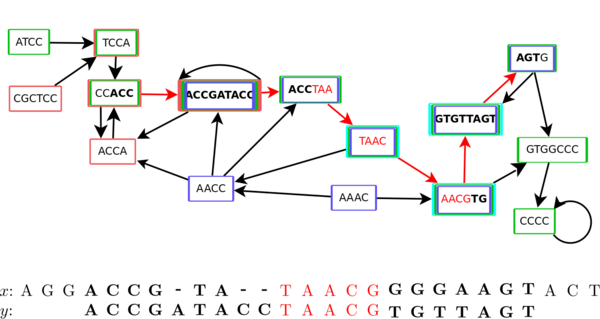
Increasing amounts of individual genomes sequenced per species motivate the usage of pangenomic approaches. Pangenomes may be represented as graphical structures, e.g., compacted colored de Bruijn graphs, which offer a low memory usage and facilitate reference-free sequence comparisons. Here we develop a new, heuristical method to find all maximum scoring local alignments between a DNA query sequence and a pangenome represented as a compacted colored de Bruijn graph. Furthermore, we introduce the notions of quorum and search color set allowing to concentrate searches on any part of the pangenome. The source code of our implementation and test data are available on gitlab.
Machine learning based analysis of crop regulatory networks
Donat Wulf (Supervisors: Bräutigam, Sczyrba)

Gene regulation is an important mechanism for organisms to react to changing environmental conditions. These regulatory mechanisms are governed by transcription factors and organized in a gene regulatory network (GRN). Machine learning enables the inference of these networks. In this project, I develop methods to establish and analyze GRNs by gene ontology enrichment tools. I compare GRNs and transcription factor binding sites by high throughput construction of phylogenetic trees and intragroup analyses. GRNs are validated by DAP-seq and EMSA.
 Except where otherwise noted, content on this wiki is licensed under the following license: CC Attribution-Share Alike 4.0 International
Except where otherwise noted, content on this wiki is licensed under the following license: CC Attribution-Share Alike 4.0 International